EDITORIAL | https://doi.org/10.5005/jp-journals-10100-6109 |
Evolving Landscape and Challenges of Autoimmune Encephalitis
1Department of Critical Care Medicine, Institute of Postgraduate Medical Education and Research and Seth Sukhlal Karnani Memorial Hospital, West Bengal, India
2Department of Medicine, Agartala Government Medical College, Tripura, India
Corresponding Author: Anup K Bhattacharya, Department of Medicine, Agartala Government Medical College, Tripura, India, e-mail: anupbhattachary@yahoo.co.in
How to cite this article Paul R, Bhattacharya AK. Evolving Landscape and Challenges of Autoimmune Encephalitis. Bengal Physician Journal 2019;6(1):1–2.
Source of support: Nil
Conflict of interest: None
Autoimmune encephalitis (AE) is a diverse group of disorders associated with immune-mediated (or more specifically antibody-mediated) attack on central nervous system (CNS) structures.1 Though earlier thought to be relatively rare, this condition is now increasingly recognized in patients with various clinical manifestations. AE is not a single clinical entity but a group of closely-related diseases with overlapping clinical and neuroimaging features.1 Previously this disease was believed to be primarily associated with an underlying malignancy. But now, antibodies against synaptic/neuronal surface proteins with varying clinical presentations are being discovered with or without any underlying malignancy.2
Antibodies causing AE are being discovered very fast. There is also an entity called seronegative AE, but this may just represent encephalitis caused by yet-undiscovered antibodies.2 The disease can present with widely varying manifestations such as subtle cognitive impairment, psychiatric manifestations, convulsions, focal neurodeficit, or movement disorders, singly or in combinations.2,3
Various antibodies are implicated in the pathophysiology of AE. There are two types of antigens against which antibodies are detected: cell surface antigens and cytoplasmic antigens. The prototype is anti-NMDA-R antibody against a neuronal cell surface antigen.3 Other similar antibodies directed against cell surface antigens include anti-AMPA-R, anti-GABAb, anti-LGI-1, anti-CASPR2, anti-glycine receptor, and anti-DPPX. Antibodies directed against intracellular antigens are mostly paraneoplastic antibodies such as anti-Hu and Yo. The non-paraneoplastic intracellular antigen against which antibody has been discovered is GAD65.
Clinical features, age of onset, gender predilection, and prognosis vary according to the type of antibody.3 For example, in the best-described prototype model of AE (the anti-NMDA-R antibody associated disease), the disease occurs at a young age (median: 20 years), mostly in females. This usually starts with non-specific symptoms such as headache, fever, and lethargy. Slowly, over days and weeks, these symptoms progresses and manifests as language problems, seizures, and/or movement disorders.3 For anti-LGI1 antibodies, features may include myoclonus, faciobrachial dystonic seizures or sleep disorders. Anti-GABAb antibodies can present with the opsoclonus-myoclonus syndrome, while anti-adenylate kinase antibodies can present with severe short term memory loss.3 Anti-GAD65 antibody, which is produced against an intracellular antigen, presents with limbic encephalitis, seizures, and cerebellar ataxia. Stiff person variant with excessive startle and head retraction reflexes are associated with glycine receptor antibodies.
When should you suspect an antibody-mediated encephalopathy?
- Temporal evolution—rapid development of symptoms.
- New onset seizures, psychosis, or cognitive decline.
- By the specific type of syndromes—faciobrachial dystonic seizure, opsoclonus-myoclonus syndrome, and stiff-person syndrome.
- A propensity to autoimmunity in the patient or family.
- The presence of a history of or suspicion of a neoplastic process.
Thus, it is evident that there is no single clinical feature that is diagnostic of the condition and early diagnosis can be challenging. The most important differential diagnosis to rule out is infectious encephalitis. The initial CSF findings in AE can be non-specific with lymphocytic pleocytosis and a mild-to-moderate increase in protein. CSF oligoclonal bands may also be found.4 But none of these are diagnostic. Diagnosis of AE requires the detection of specific antibodies in CSF and/or serum. Sometimes, serum antibodies may be undetectable, but CSF antibodies are always found. The only exception is anti-LGI1 syndrome, where serum antibody may be more common than CSF antibody.5 Antibody titers may be related with disease severity, but data on that correlation are still inadequate. Usually, the IgG antibody titer is checked. IgA or IgM has doubtful significance.
One important controversy surrounding antibody detection is the case of anti-VGKC antibody.6 This was one of the first antibodies found to be associated with AE. But later, it was shown that anti-VGKC has significance only when associated with anti-LGI1 or anti-CASPR2.6 In an article published in Neurology in 2016, Van Sonderen et al. concluded that only anti-VGKC antibody measurement has no clinical significance.6
CLASSIC PRESENTATION OF ANTI-NMDAR ENCEPHALOPATHY
Prodrome (agitation, psychosis, and memory deficit), abnormal movements, coma, and dysautonomia (with or without seizure).
CLINICAL FEATURES OF VGKC (LGI1 CASPR2) AB ENCEPHALITIS
- Recent onset chorea or isolated movement disorder.
- LGI1-related faciobrachial dystonia, episodic bradycardia/drop attacks, and hyponatremia.
- CJD-like presentation with myoclonus, ataxia, and painful neuropathy.
- Morvan’s fibrillary chorea (thymoma).
In case of progressive encephalomyelopathy with rigidity (axial/limb), opsoclonus-myoclonus syndrome search for occult neoplasm is warranted.
IgLON5 antibody (a neuronal cell adhesion protein) is associated with prominent REM and non-REM sleep disorder, central hypoventilation, ataxia, and dysautonomia. In young children, another entity to consider in cases of encephalitis of uncertain etiology is Rasmussen encephalitis. But that discussion is beyond the scope of this article.
MRI changes in the brain and/or spinal cord in AE are varied. It can be completely normal in some cases also. Usual MRI changes include T2/FLAIR hyperintensities. Any part of the cortex may be involved, but usual sites are the temporal lobe, insular cortex, or cingulate gyrus.7 But, especially in anti-NMDA-R encephalitis, imaging findings may be absent to start with. In such cases, if clinical suspicion for AE is high, then FDG-PET of the brain should be done.7
The EEG can also be abnormal.3 There can be generalized slowing, PLED, or delta brush.3
Treatment for AE is still not standardized. Common treatment options include IVIg, corticosteroids, or plasma exchange. These can also be used in combination. Young patients generally respond better compared to the elderly. If the first line-therapies mentioned above do not work, then in the second line, rituximab or cyclophosphamide may be used. If steroids lead to initial improvement, then later steroid-sparing agents such as mycophenolate or azathioprine may be used. In a study from Austria, published in 2018, it was seen that very few patients with AE responded to the first-line therapy alone and in most cases, escalation to the second-line therapy was needed.8 There is no clear guideline till now8 regarding the usage of second-line therapy.
Autoimmune encephalitis is thus a disease with varying clinical manifestations. Diagnosis of this condition requires a high index of clinical suspicion. Treatment is done with various immunomodulatory drugs but there are no clear guidelines, and clinicians have to modify the therapy based on clinical judgment.
As the spectrum of autoimmune enchaphalits is a rapidly developing with more and more number of antibodies being detected each day with its varying clinical presentations, physicians should keep themselves updated to keep pace with new developments for the benefit of patients and early intervention with immunomodulators, leading to a good clinical outcome.
PATTERN OF ANTIGENS, THEIR ASSOCIATION, AND RESPONSE TO TREATMENT
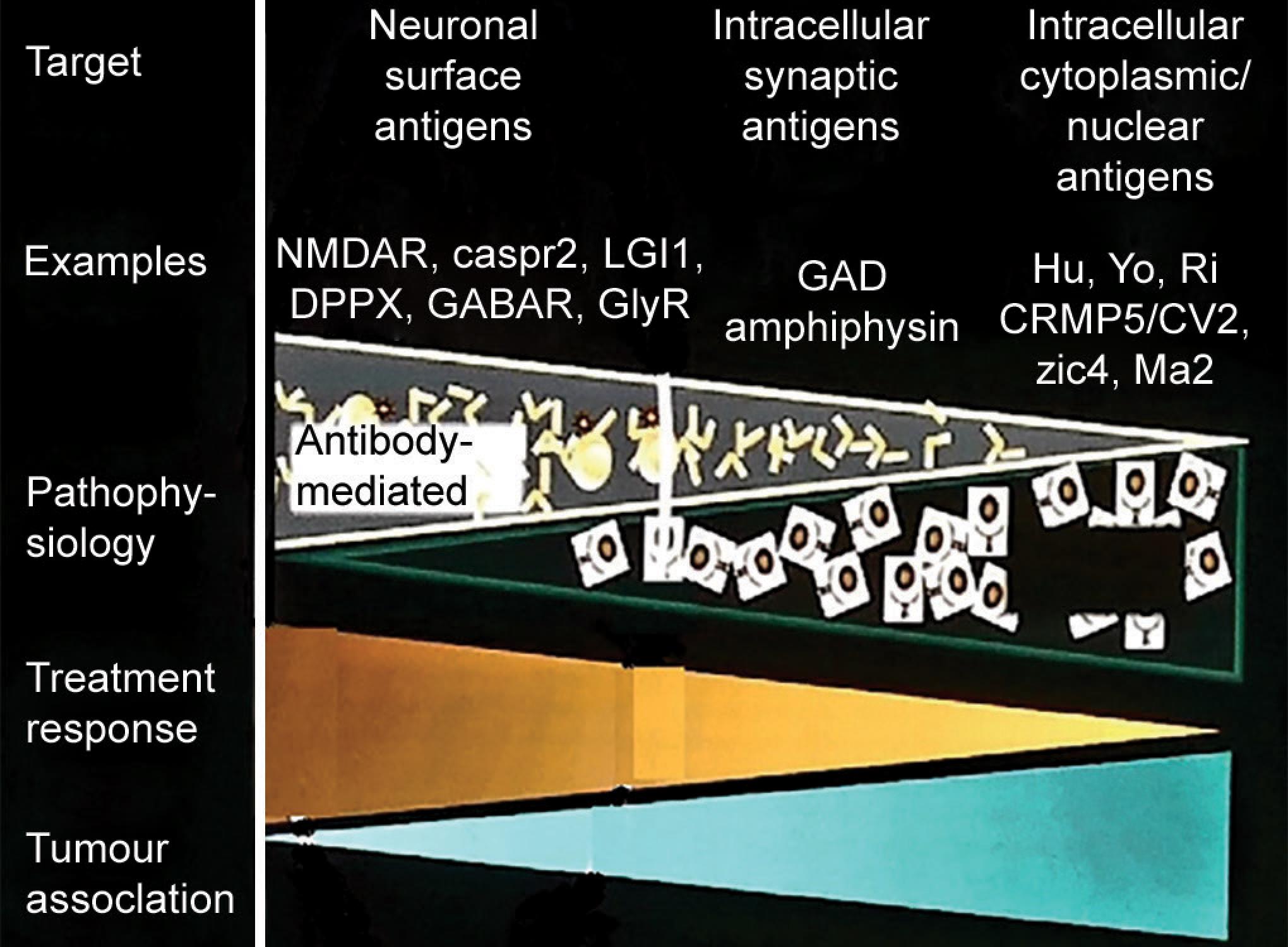
This diagram is taken from lecture notes of Professor Kailash P Bhatia at the 3D approach to movement disorder at Chennai on Aug 2019.
REFERENCES
1. Kelley BP, Patel SC, et al. Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol 2017;38:1070–1078. DOI: 10.3174/ajnr.A5086.
2. Blaabjerg M, Seifert-Held T, et al. Editorial: emerging challenges in the diagnosis and treatment of autoimmune encephalitis. Front Neurol 2019;10:146. DOI: 10.3389/fneur.2019.00146.
3. Hermetter C, Fazekas F, et al. Systematic review: syndromes, early diagnosis, and treatment in autoimmune encephalitis. Front Neurol 2018;9:706. DOI: 10.3389/fneur.2018.00706.
4. Malter MP, Elger CE, et al. Diagnostic value of CSF findings in antibody-associated limbic and anti-NMDAR-encephalitis. Seizure 2013;22:136–140. DOI: 10.1016/j.seizure.2012.12.013.
5. Graus F, Titulaer MJ, et al. Clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404. DOI: 10.1016/S1474-4422(15)00401-9.
6. Van Sonderen A, Schreurs M, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology 2016;86:1692–1699. DOI: 10.1212/WNL.0000000000002637.
7. Kelley BP, Patel SC, et al. Autoimmune Encephalitis: Pathophysiology and Imaging Review of an Overlooked Diagnosis. AJNR Am J Neuroradiol 2017;38:1070–1078. DOI: 10.3174/ajnr.A5086.
8. Macher S, Zimprich F, et al. Management of autoimmune encephalitis: an observational monocentric study of 38 patients. Front Immunol 2018;9:2708. DOI: 10.3389/fimmu.2018.02708.
________________________
© The Author(s). 2019 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.