CASE REPORT |
https://doi.org/10.5005/jp-journals-10070-7013 |
Isoniazid-Induced Encephalopathy in a Chronic Kidney Disease Patient: A Case Report
1,3,4Department of Internal Medicine, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh
2Noakhali 250 Bed General Hospital, Noakhali, Chattogram, Bangladesh
Corresponding Author: Shohael M Arafat, Department of Internal Medicine, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh, Phone: +880 1732330313, e-mail: arafat2001@gmail.com
How to cite this article: Alam MA, Uddin MJ, Khan AH, et al. Isoniazid-induced Encephalopathy in a Chronic Kidney Disease Patient: A Case Report. Bengal Physician Journal 2020;7(1):14–16.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Drug-induced encephalopathy is a well-known side effect of many drugs. Isoniazid (INH), a first-line drug used in the treatment of tuberculosis, can cause encephalopathy in chronic kidney disease (CKD) patients though other isoniazid-related neurotoxicities are more commonly encountered in general population. We report isoniazid-induced encephalopathy in a female patient with CKD. She has been given rifampicin, INH, pyrazinamide, and ethambutol with pyridoxine and prednisolone for the treatment of Pott's disease of cervical spine. The patient developed recurrent episodes of altered consciousness following the treatment. After exclusion of other causes, isoniazid-induced encephalopathy was suspected, which was further proved by symptomatic improvement after stopping isoniazid.
Keywords: Chronic kidney disease, Isoniazid-induced encephalopathy, Tuberculosis.
INTRODUCTION
Being one of the top ten causes of death worldwide, tuberculosis (TB) remains to be a major cause of ill health globally. According to Global Tuberculosis Report 2019, an estimated 10 million people were infected with TB and 1.2 million HIV-negative people died of TB in 2018.1
In patients with chronic kidney disease (CKD), the incidence of tuberculosis is 10-fold greater than that of the normal population due to reduced cellular immunity resulting from uremia.2,3
Furthermore, incidences of side effects due to anti-TB drug treatment are more frequently reported in CKD patients.4
Isoniazid, also known as isonicotinic acid hydrazide (INH), is an antibiotic used as one of the first-line drugs in the treatment of tuberculosis. The level of INH in the plasma depends on the genetically determined slow or fast acetylation process in the liver. Since the route of excretion of INH is primarily through kidneys in both free and acetylated forms, the dose of INH may need to be reduced in patients with CKD.5
The neurotoxic side effects associated with INH use are well recognized. These may range from peripheral neuropathy, optic neuritis, encephalopathy, ataxia, dysarthria, and seizure to psychiatric manifestations such as anxiety, depression, paranoia, schizophreniform psychosis, obsessive–compulsive disorder, and mania.6
Mechanism of these effects is inhibition of the phosphorylation of pyridoxine, causing decreased production of pyridoxal-5-phosphate, which is a coenzyme involved in neurotransmission.3
Moreover, decreased acetylation of INH in patients with CKD contributes to a longer half-life of the drug.7
In 1961, Castaigne et al. described a case of INH-induced encephalopathy, which was one of the earliest drug-induced encephalopathies reported in the literature.8
Patients receiving dialysis and slow acetylators conferred by NAT2 polymorphisms have more risk of developing INH encephalopathy.9
Here, we are presenting a case of encephalopathy in a CKD patient caused by administration of isoniazid for the treatment of Pott's disease of cervical spine.
CASE DESCRIPTION
This 55-year-old Bangladeshi woman, a known case of hypertension, hypothyroidism, diabetes mellitus, and chronic kidney disease, presented with neck pain and low-grade fever. She was diagnosed as a case of Pott's disease of cervical spine on the basis of clinical scenario, X-ray (Fig. 1) and MRI of cervical spine (Fig. 2), and other laboratory evidences.
She has been treated with isoniazid 225 mg/day, rifampicin 450 mg/day, ethambutol 400 mg, pyrazinamide 100 mg, pyridoxine 20 mg, and prednisolone 60 mg after necessary adjustment for impaired renal function. At that time, her serum creatinine was 6.03 mg/dL and surgical intervention in the cervical spine was planned. But after 3 days of treatment, patient became unconscious with estimated Glasgow Coma Score 6. At this point, her serum creatinine (7 mg/dL) and blood urea (218 mg/dL) level were found to be high. Serum electrolyte and RBS were within normal range. She was promptly shifted to ICU where her SGPT, serum albumin, and serum calcium were found to be normal but serum potassium level was mildly raised (5.5 mmol/L). Arterial blood gas analysis revealed metabolic acidosis, complete blood count showed high ESR (98 mm in first hour) and neutrophilia (80%) with leukocyte count (10,600/mm3) at the upper limit of normal value, and urine routine examination showed proteinuria. Anti-TB therapy was stopped immediately. Adrenal insufficiency was excluded by normal ACTH and cortisol level. She was given three sessions of hemodialysis through right femoral venous catheter. She gained full consciousness after 7 days from the first day of hemodialysis. Anti-TB therapy was restarted from the next day with the same previous regimen. She was discharged 7 days after starting therapy during which her serum creatinine was 4.6 mg/dL. She was advised to follow-up after 1 month. But the very next day of discharge, patient complained of anorexia and excessive sleepiness. Three days later, the patient had to get admitted to hospital again due to confusion and breathlessness. On examination, she appeared pale and edematous with puffy face and Glasgow coma score was 8. She received another session of hemodialysis the next day along with sodium bicarbonate and furosemide. She was shifted to ICU again as her condition remained unchanged. Her ABG showed that metabolic acidosis, serum urea (225 mg/dL), serum phosphate (8.7 mg/dL), and serum creatinine (9.5 mg/dL) were also found to be high, whereas serum ammonia (18 μmol/L), serum electrolyte, liver enzymes, and prothrombin time were normal. Blood culture yielded no growth, but urine culture yielded growth of ESBL-positive E. coli. Another session of hemodialysis was given, and anti-TB therapy was stopped. Patient was shifted to general ward after 4 days, but she was still drowsy with GCS 12. In 11 days, she was fully alert and oriented. In the meantime, CT-guided FNAC from C5 vertebral body revealed tubercular granulomatous inflammation (Fig. 3), which impelled us to restart anti-TB therapy again.

Fig. 1: X-ray of cervical spine
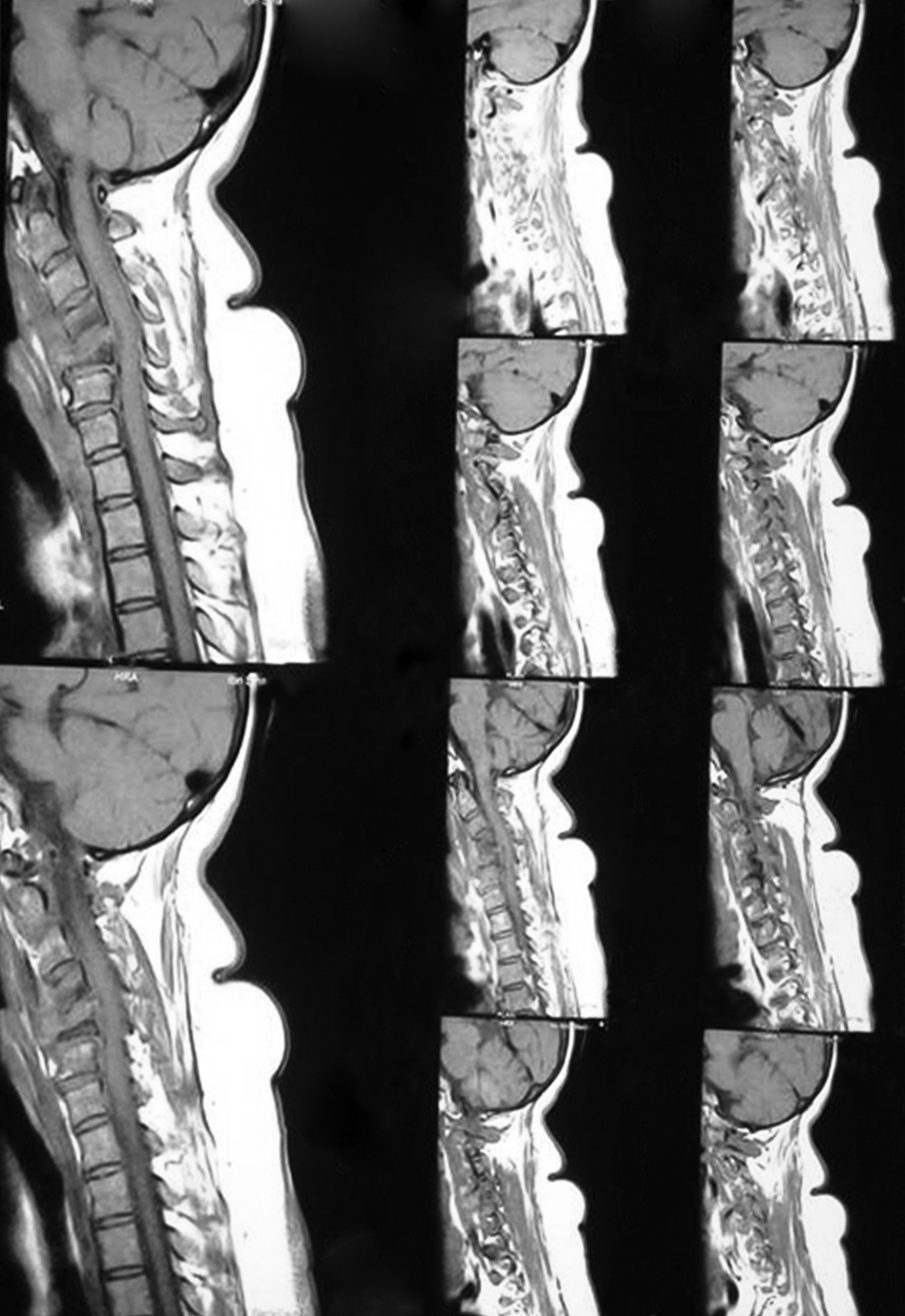
Fig. 2: MRI of cervical spine
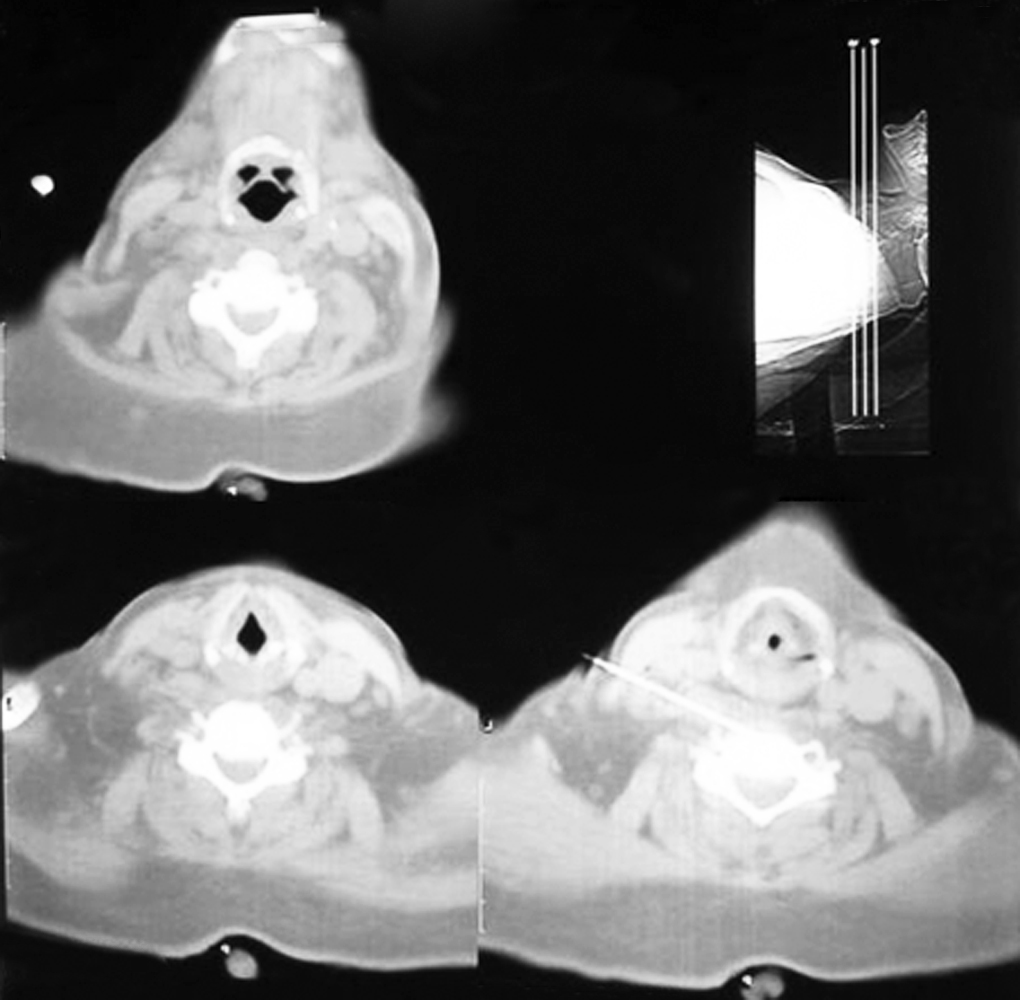
Fig. 3: CT-guided FNAC (C5 vertebral body)
As a temporal relationship between anti-TB therapy and recurrent unconsciousness was anticipated, decision was taken to reintroduce anti-TB drugs one by one in incremental doses. Isoniazid was the first drug to be reintroduced, followed by ethambutol. But both the drugs were omitted as the patient became drowsy on the seventh day of starting isoniazid (and second day of ethambutol). Since the patient's orientation improved, pyrazinamide was reintroduced after 7 days, followed by rifampicin and ethambutol again along with prednisolone. Expected full dose of anti-TB therapy was restarted within 1 month, which consisted of rifampicin, ethambutol, pyrazinamide, and prednisolone. There was no incidence of unconsciousness or drowsiness since then and no side effects identified in the subsequent follow-ups. No hemodialysis required during anti-TB therapy though A-V fistula was constructed with the aim of long-term renal replacement therapy. Her last serum creatinine was 6.01 mg/dL.
DISCUSSION
Isoniazid-induced encephalopathy is basically diagnosed clinically by the exclusion of other infective causes or metabolic disturbances. In this patient, the diagnosis is strongly supported by temporal relationship between the onset of altered consciousness and the initiation of INH therapy (4–14 days), completed recovery (within 1 week) after cessation of the drug, and reproducibility of symptoms on reintroduction of the drug. The clinical features and investigation workups render the diagnosis of other conditions like septicemia, electrolyte imbalance, hypertensive encephalopathy, Wernicke's encephalopathy, aluminum toxicity, uremic encephalopathy, hepatic encephalopathy, acute disequilibrium syndrome, and cerebrovascular diseases unlikely.
INH treatment may increase the risk of developing pellagra by reducing the conversion of tryptophan to niacin. Pellagra psychosis may be mistaken for INH-induced encephalopathy where typical dermatitis or diarrhea is absent.10 However, our patient showed complete resolution of symptoms on stopping INH only and no niacin replacement needed, which made the diagnosis of pellagra less likely.
Though plasma levels of INH and acetylator status were not determined in our patient, her Asiatic origin does not render her to fall under slow acetylator phenotype.
The mechanism of INH-induced encephalopathy is still obscured. It may be caused by INH-induced deficiency of pyridoxine and pyridoxal phosphate, which act as the coenzymes for the production of synaptic transmitters.6
Predisposing factors for developing neuropsychiatric complications of INH include old age, diabetes, hepatic insufficiency, alcoholism, malnourishment, hyperthyroidism, slow acetylators, brain damage, past or family history of psychiatric illness, use of high dose of INH (>5 mg/kg daily), and concomitant administration of monoamine oxidase inhibitors.6
INH-induced encephalopathy can be developed any time between first few days to 5 months after initiation of INH therapy. On the contrary, recovery is more predictable, usually within 1 week of stopping of INH.11
In spite of using prophylactic pyridoxine in our patient, INH-induced encephalopathy occurred. There are case reports that describe the development of encephalopathy and cerebellar syndrome in patients with CKD while on regular pyridoxine.12 So, the role of pyridoxine in preventing isoniazid-induced encephalopathy is uncertain though the useful effect of pyridoxine on INH-induced peripheral neuropathy is well established.6
Nevertheless, some authorities recommend a higher dose of pyridoxine to prevent INH-induced encephalopathy in CKD patients.8
CONCLUSION
In conclusion, isoniazid-induced encephalopathy should be kept in mind when patients with CKD under treatment with INH develop confusion. Early recognition of such diagnosis can be crucial because cessation of the drug can result in complete recovery of the patient.
REFERENCES
1. World Health Organization (WHO). Global tuberculosis report 2019. Geneva, Switzerland: World Health Organization; 2019. Available from: https://www.who.int/teams/global-tuberculosis-programme/tb-reports. ISBN: 9789241565714
2. Quantrill SJ, Woodhead MA, Bell CE, et al. Side-effects of antituberculosis drug treatment in patients with chronic renal failure. Eur Respir J 2002;20(2): 440–443.DOI: 10.1183/09031936.02.00298002.
3. Siskind MS, Thienemann D, Kirlin L. Isoniazid-induced neurotoxicity in chronic dialysis patients: report of three cases and a review of the literature. Nephron 1993;64(2): 303–306.DOI: 10.1159/000187331.
4. Hussein MM, Mooij JM, Roujouleh H. Tuberculosis and chronic renal disease. Semin Dial 2003;16(1): 38–44.DOI: 10.1046/j.1525-139x.2003.03010.x.
5. Abbas MT, Khan FY, Sulimon S, et al. Encephalopathy secondary to isoniazid in patients on hemodialysis. Indian J Nephrol 2013;23(1): 54–56.DOI: 10.4103/0971-4065.107206.
6. Cheung WC, Lo CY, Lo WK, et al. Isoniazid induced encephalopathy in dialysis patients. Tuber Lung Dis 1993;74(2): 136–139.DOI: 10.1016/0962-8479(93)90042-V.
7. Kim YG, Shin JG, Shin SG, et al. Decreased acetylation of isoniazid in chronic renal failure. Clin Pharmacol Ther 1993;54(6): 612–620.DOI: 10.1038/clpt.1993.198.
8. Haque WM, Samad T, Rahim MA, et al. Drug induced encephalopathy in patients with chronic kidney disease: a case series. BIRDEM Med J 2018;8(2): 172–176.DOI: 10.3329/birdem.v8i2.36650.
9. Constantinescu SM, Buysschaert B, Haufroid V, et al. Chronic dialysis, NAT2 polymorphisms, and the risk of isoniazid-induced encephalopathy—case report and literature review. BMC Nephrol 2017;18(1):282. DOI: 10.1186/s12882-017-0703-6.
10. Ishii NO, Nishihara Y. Pellagra encephalopathy among tuberculous patients: its relation to isoniazid therapy. J Neurol Neurosurg Psychiatry. 1985;48(7): 628–634.DOI: 10.1136/jnnp.48.7.628.
11. Yih WH, Chun CC, Ming CY, et al. Encephalopathy caused by isoniazid in a patient with end stage renal disease with extrapulmonary tuberculosis. Ren Fail 2003;25(1): 135–138.DOI: 10.1081/jdi-120017476.
12. Blumberg EA, Gil RA. Cerebellar syndrome caused by isoniazid. DICP 1990;24(9): 829–831.DOI: 10.1177/106002809002400906.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.