CASE REPORT |
https://doi.org/10.5005/jp-journals-10070-7039 |
Ischemic Cardiomyopathy in a Case of Takayasu’s Arteritis
1–6Department of General Medicine, Calcutta National Medical College and Hospital, Kolkata, West Bengal, India
Corresponding Author: Sujoy Sarkar, Department of General Medicine, Calcutta National Medical College and Hospital, Kolkata, West Bengal, India, Phone: +91 9831570443, e-mail: sujoysarkarcmc@gmail.com
How to cite this article: Mukherjee S, Samanta S, Saha U, et al. Ischemic Cardiomyopathy in a Case of Takayasu’s Arteritis. Bengal Physician Journal 2021;8(1):17–20.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Takayasu’s arteritis (TA) is a rare form of granulomatous vasculitis of unknown etiology. It is also known as “pulseless disease” or “aortic arch syndrome” and commonly occurs in young- or middle-aged females. We describe a 28-year-old female who presented with chest pain and dyspnea in the Department of Medicine of Calcutta National Medical College and Hospital, Kolkata, West Bengal, India. On initial examination, the patient had absent pulses in all four limbs, and the patient’s blood pressure (BP) was not recordable. Clinical examination also revealed features of biventricular failure. Relevant routine investigations like electrocardiogram (ECG), two-dimensional Doppler electrocardiography (2D-Echo), cardiac markers, and blood parameters were done. 2D-Echo was suggestive of ischemic cardiomyopathy. Considering the patient’s physical findings along with raised erythrocyte sedimentation rate (ESR), further investigations like CT angiography (CTA) were done, which led to the diagnosis of TA. The patient was treated with oral steroid, diuretic, beta-blocker, and antiplatelet and then discharged in a stable condition.
Keywords: Heart failure, Steroid, Vasculitis.
INTRODUCTION
Takayasu’s arteritis (TA) is a chronic inflammatory and stenotic disease of medium- and large-sized arteries with a strong predilection for the aorta and its major branches, the specific cause of which is not known. It is an uncommon disease with an estimated annual incidence rate of 1.2–2.6 cases per million. It is most prevalent in adolescent girls and young women.1 Though exact cause or pathogenesis is not known, studies have shown that T cell, B cell, and macrophages after getting activated by some trigger factors play an important role in inflammation, narrowing, and aneurysm of blood vessels.2 It is one of the commonest causes of renovascular hypertension in India.3 Here, we present a rare presentation of TA in which the patient presented with symptoms and signs of cardiac ischemia and heart failure. This case report emphasizes the need for detailed physical examination and investigations and a high index of suspicion for the diagnosis of this disease.
CASE DESCRIPTION
History
A 28-year-old female, mother of a child, presented with intermittent exertional left-sided chest pain for 2 months and progressive dyspnea for 1 month. The patient’s chest pain was typical anginal in nature, started on exertion, and was relieved by taking rest. The patient developed gradual onset and progressive dyspnea for 1 month. It was initially New York Heart Association (NYHA) class I; but within 1-month period, it became NYHA class IV. The patient also complained of bilateral pedal swelling for 15 days. This swelling was pitting in nature. There was no history of fever, joint pains, cough, hemoptysis, headache, syncope, visual disturbances, or limb claudication pain.
There was no history of rheumatic fever, tuberculosis, blood transfusion, any surgery, and high-risk behavior.
On Examination
On examination, the patient was conscious, alert, cooperative, and well oriented to time, place, and person. The patient was of average built with no signs of malnutrition. During the examination of pulse, it was found that all the peripheral pulses were absent, except bilateral carotid artery pulsation that too was feeble, and her blood pressure (BP) could not be measured. The respiratory rate was 28 per minute and thoracoabdominal in nature. Pallor, cyanosis, clubbing, and icterus were absent, but bilateral pitting pedal edema was present. Jugular venous pressure was raised 10 cm from the sternal angle, pulsatile in character. Cardiovascular system examination revealed a normally positioned apex with audible LV S3, no other added sound or murmur. Bibasal fine crepitations were also present.
Blood investigations showed Hb—10.2 g/dL, TLC—10,800/cumm, neutrophil 56%, lymphocyte 40%, eosinophil 3%, monocyte 1%, ESR—65 mm at first hour. CRP—20.63 µg/mL.
ECG showed ST depression with T wave inversion in leads V3 to V6. Trop T was negative.
CPK—100.0 U/L, CPK-MB—6.0 U/L. ANA profile including ANA, dsDNA, anti-Smith, and APLA profile all were negative. HIV 1 and 2, HBs Ag, anti-HCV, and VDRL were also negative.
2D-Echo showed normal LA, LVIDd 58 mm, LVIDs 48 mm, mild mitral regurgitation, global hypokinesia, and LVEF = 30% as well as grade I diastolic dysfunction, no vegetation, and no pericardial effusion. CTA of the arch of the aorta revealed diffuse thickening of ascending, descending, and arch of aorta with significant luminal narrowing involving origins, intrathoracic, and visualized cervical course of the brachiocephalic, left common carotid, and left subclavian arteries (Fig. 1). CTA of the abdominal aorta showed significant narrowing at the origin of the celiac artery, superior mesenteric artery, inferior mesenteric artery, and right renal artery (Fig. 2). Magnetic resonance (MR) angiography of the brain revealed nonvisualization of right internal carotid artery up to C4 segment, decreased caliber of left internal carotid artery C2, C3, and C4 segments, and prominent bilateral posterior communicating arteries (Fig. 3).
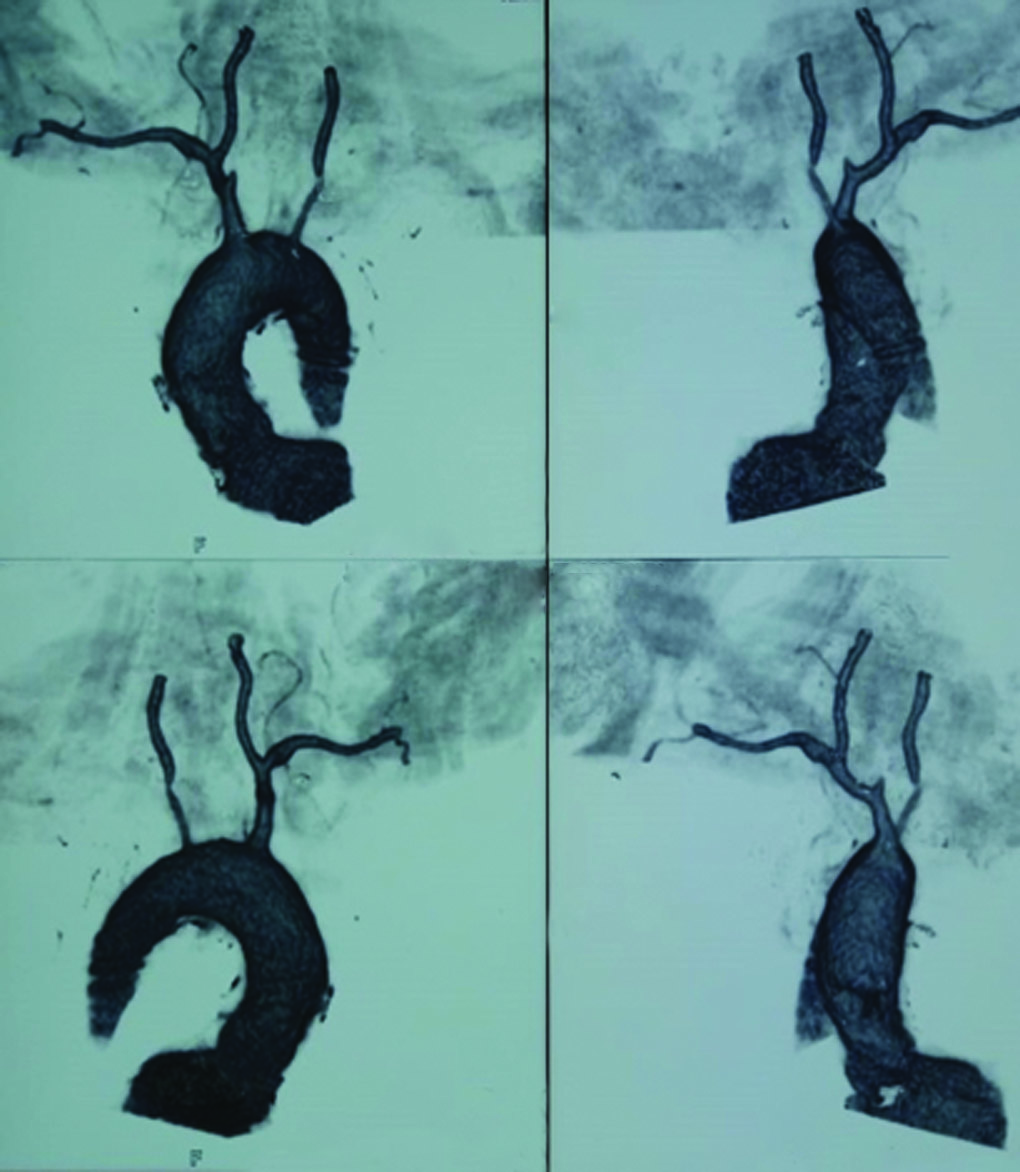
Fig. 1: CT angiography of the arch of aorta showing diffuse thickening of ascending, descending, and arch of aorta associated with significant luminal narrowing of brachiocephalic, left common carotid, and left subclavian arteries

Fig. 2: CT angiography of abdominal aorta showing significant narrowing at the origin of branches of the abdominal aorta
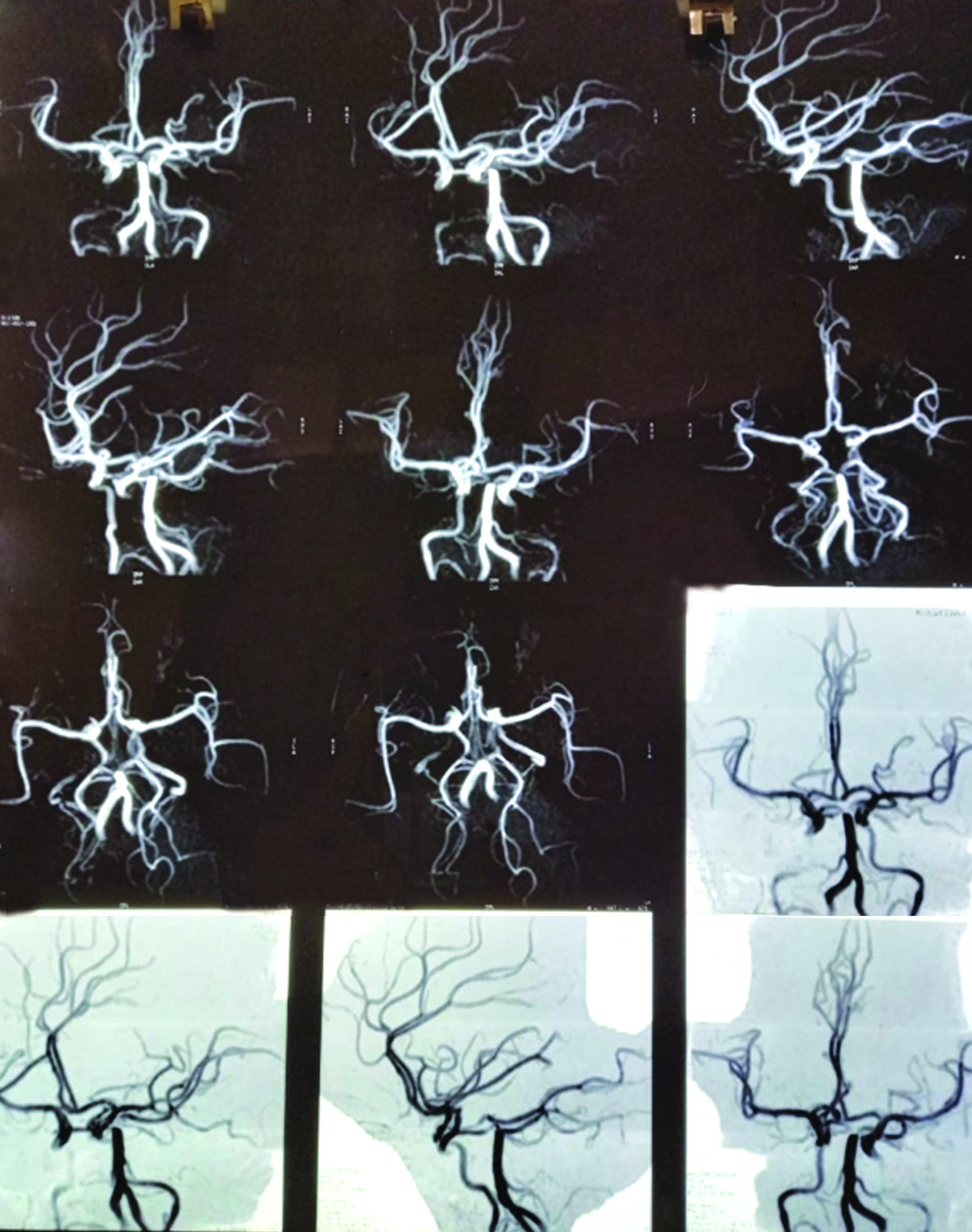
Fig. 3: MR angiography brain showing nonvisualization of the right internal carotid artery, decreased caliber of the left internal carotid artery, and prominent bilateral posterior communicating arteries
This case was diagnosed as type V TA with ischemic cardiomyopathy.
The patient was treated with oral steroid (tab prednisolone 1 mg/kg), diuretic, beta-blocker, and antiplatelet. The patient’s clinical condition improved remarkably, and then the patient was discharged in a clinically stable condition.
DISCUSSION
TA is a rare condition primarily affecting large and medium blood vessels, characterized by granulomatous inflammation leading to thickening of the vessel wall, fibrosis, stenosis, and thrombosis. Aorta and its branches are predominantly affected. The etiology remains unknown. Symptoms of the disease depend on end-organ ischemia.
TA progresses through three stages; however, in actual practice, most patients actually do not fall in such groupings.
- First stage: Early systemic stage or prevasculitic stage, when the patient complains of constitutional symptoms.
- Second stage: Vascular inflammatory stage.
- Third stage: Burnt-out stage.
Classification based on the site of involvement.4
- Type I: Aortic arch.
- Type II: Descending thoracic aorta.
- Type III: Descending thoracic aorta and abdominal aorta.
- Type IV: Abdominal aorta only.
- Type V: Aortic arch, descending thoracic aorta, and abdominal aorta.
The first diagnostic criteria for TA was developed by Ishikawa in 1988, which was modified by Sharma et al.5—the American College of Rheumatology 1990 criteria6:
- Age of onset <40 years.
- Claudication of extremities.
- Decreased pulsation of one or both brachial arteries.
- Systolic BP difference of at least 10 mm Hg in between arms.
- Bruit over one or both subclavian arteries and aorta.
- Aortogram abnormalities.
At least three of the above six criteria have to be met.
TA is one of the commonest causes of renovascular hypertension in India.3 The etiology of TA is unknown, but an association with tuberculosis has been reported.7 Coronary arteries can also be involved in TA and can be divided into three types based on histopathology.8,9 Type I represents stenosis or occlusion of the coronary vessels. Type II is arteritis of the coronary vessels, whereas type III includes coronary aneurysms. Cardiac ischemia leading to dilated cardiomyopathy and heart failure in cases of TA is relatively rare and reported in only around 6% of cases.10
Steroids are the mainstay of treatment of TA. Tocilizumab, anti-IL6 receptor antibody, produces clinical response and has steroid-sparing effects in patients with refractory TA.11 Rituximab, a chimeric anti-CD20 antibody, has been shown to improve clinical signs and symptoms of TA.12 Cytotoxic agents like tacrolimus and mycophenolate mofetil are also used mainly in patients with steroid-resistant or relapsing disease.13 Surgical therapeutic measures include angioplasty or surgical revascularization procedures undertaken during periods of disease remission.
CONCLUSION
TA is a rare but fatal disorder if not diagnosed and treated. High index of clinical suspicion with detailed clinical examination and history taking remains the key to diagnosis. Hence, in all female patients of child-bearing age, presenting with angina, dyspnea, or heart failure, the diagnosis of TA should be kept in mind.
REFERENCES
1. American College of Physicians (ACP). Systemic vasculitis. Medical knowledge self-assessment program (MKSAP-15): rheumatology. Philadelphia, PA: ACP, 2009, pp. 65–67.
2. Espinoza JL, Ai S, Matsumura I. New insights on the pathogenesis of Takayasu arteritis: revisiting the microbial theory. Pathogens. 2018;7:73. DOI: 10.3390/pathogens7030073.
3. Arora P, Kher V, Singhal MK, et al. Renal arterystenosis in nonspecific aortoarteritis: spectrum of disease in children and adults. Kidney Blood Press Res 1997;20:285–289. DOI: 10.1159/000174160.
4. Jain S, Kumari S, Ganguly NK, et al. Current status of Takayasu Arteritis in India. Int J Cardiol 1996; 54(Suppl): S111–S116. DOI: 10.1016/s0167-5273(96)88780-8.
5. De Souza AW, de Carvalho JF. Diagnostic and classification criteria of Takayasu arteritis. J Autoimmun 2014;48–49:79–83. DOI: 10.1016/j.jaut.2014.01.012.
6. Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum 1990;33(8):1129–1134. DOI: 10.1002/art.1780330811.
7. Duzova A, Turkmen O, Cinar A, et al. Takayasu arteritis and tuberculosis: a case report. Clin Rheumatol 2000;19:486–489. DOI: 10.1007/s100670070013.
8. Sun T, Zhang H, Ma W, et al. Coronary artery involvement in Takayasu arteritis in 45 Chinese patients. J Rheumatol 2013;40:493–497. DOI: 10.3899/jrheum.120813.
9. Matsubara O, Kuwata T, Nemoto T, et al. Coronary artery lesions in Takayasu arteritis: pathological considerations. Heart Vessels Suppl 1992;7:26–31. DOI: 10.1007/bf01744540.
10. Ghosh S, Sinha DP, Ghosh S, et al. Dilated cardiomyopathy in non specific aortoarteritis. Indian Heart J 1999;51:527–531. PMID: 10721644
11. Nakaoka Y, Isobe M, Takei S, et al. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann Rheum Dis 2018;77(3):348–354. DOI: 10.1136/annrheumdis-2017-211878.
12. Unizony S, Stone JH, Stone JR. New treatment strategies in large-vessel vasculitis. Curr Opin Rheumatol 2013;25(1):3–9. DOI: 10.1097/BOR.0b013e32835b133a.
13. Yokoe I, Haraoka H, Harashima H. A patient with Takayasu’s arteritis and rheumatoid arthritis who responded to tacrolimus hydrate. Intern Med 2007;46(22):1873–1877. DOI: 10.2169/internalmedicine.46.0211.
________________________
© The Author(s). 2021 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.